Unlocking the Genetic Blueprint: Navigating the Hereditary Risk Matrix of Age-Related Macular Degeneration
Age-related macular degeneration (AMD) is the leading cause of irreversible, late-onset blindness in high-income nations. Clinically, the disease is characterized by the degeneration of the macula, the specialized region of the central retina responsible for high-acuity color vision and sharp central perception. Pathologically, AMD progresses from early stages, characterized by the accumulation of extracellular protein-lipid deposits (drusen) beneath the retinal pigment epithelium (RPE) along Bruch’s membrane (BrM), to advanced late-stage phenotypes. Late-stage AMD is bifurcated into dry AMD, characterized by geographic atrophy (GA) of the macular photoreceptors and RPE, and wet AMD, characterized by choroidal neovascularization (CNV) wherein aberrant blood vessels breach Bruch's membrane to cause hemorrhage, exudation, and rapid fibrotic scarring.

For decades, clinical management was restricted to reactive, lifestyle-driven modifications, such as smoking cessation, ultraviolet light protection, and dietary supplementation with high-dose antioxidants and zinc. However, modern ophthalmic genomics has demonstrated that advanced AMD is one of the most highly heritable conditions in medicine, with genetic variations accounting for approximately 70% of the overall disease risk. By looking beyond generic therapeutic protocols to examine the specific genetic blueprints that dictate baseline cellular vulnerability, clinical ophthalmology is entering an era of proactive, targeted vision preservation. This comprehensive review synthesizes the molecular mechanisms driving AMD heritability, focusing on the complement factor H (CFH) locus, the chromosome 10q26 (ARMS2/HTRA1) block, cumulative polygenic risk scores (PRS), protective alleles, and the modulating effects of epigenetics and the circulating metabolome.

The CFH Variant: Mechanics of Alternative Complement Overdrive
The Complement Factor H (CFH) gene, located on chromosome 1q23-32 within the Regulator of Complement Activation (RCA) gene cluster, represents one of the strongest susceptibility loci in macular degeneration. The alternative complement pathway acts as a highly active, self-amplifying arm of the innate immune system designed to neutralize invading pathogens, clear cellular debris, and coordinate local inflammatory cascades. Under physiological conditions, the CFH protein circulating in the blood acts as the primary soluble inhibitor of this alternative pathway, safeguarding healthy host tissues from accidental bystander immune damage.

The most prominent risk variant within this gene is the single-nucleotide polymorphism (SNP) rs1061170 (a 1277T→C transition in exon 9), which causes a tyrosine-to-histidine substitution at amino acid position 402 (Y402H) within the seventh short consensus repeat (SCR7) domain of the CFH protein. Carrying a single copy of the 402H risk allele (heterozygous TC genotype) increases the risk of developing AMD by a factor of 2.2 to 4.6, whereas carrying two copies (homozygous CC genotype) elevates the relative risk 5- to 7-fold compared to the low-risk TT genotype.
│
CFH binds CRP, Heparin, & Bruch's Membrane
│
Alternative Pathway C3 Convertase Inhibited
│
Debris Cleared without Bystander Inflammation
│
(Retinal Homeostasis)
VS.
│
Disrupted Binding to Polyanions & Lipids
│
Unchecked C3 Convertase & MAC Assembly
│
Impaired Debris Clearance + Low-Grade Inflammation
│
(Basal Laminar Deposits & Drusen)

The Molecular Mechanism of Y402H-Mediated Complement Dysregulation
The CFH protein is composed of 20 short consensus repeat (SCR) domains. While the complement-regulating, cofactor, and decay-accelerating activities are mediated primarily by domains SCR1–4, the SCR7 domain is responsible for anchoring the regulatory protein to polyanions on host cell surfaces and extracellular matrices. Specifically, SCR7 mediates CFH binding to heparin, glycosaminoglycans, C-reactive protein (CRP), Bruch's membrane, malondialdehyde (MDA) epitopes, and oxidized phospholipids.

The Y402H substitution alters the electrostatic charge distribution of the SCR7 binding pocket, reducing the affinity of the CFH-402H variant for these protective polyanionic ligands. Consequently, the mutant CFH protein fails to anchor properly to Bruch's membrane and the RPE surface. This is clinically supported by immunohistochemical costaining of drusen, which reveals that spheroid-like particles within drusen contain both CRP and CFH in individuals with the low-risk TT (Y402) genotype, but show a significant reduction in costaining in patients harboring the homozygous CC (H402) risk genotype.

When CFH fails to bind to CRP and Bruch's membrane, its negative regulatory control is lost. The alternative pathway C3 convertase (C3bBb) forms unchecked, triggering a self-amplifying cascade that cleaves C3 into C3a and C3b.C3b acts as an opsonin, flagging cells for phagocytosis, while also driving the downstream assembly of the membrane attack complex (MAC, or C5b-9). The accumulation of MAC within the sub-RPE space induces localized cell lysis, choriocapillaris degeneration, and Bruch's membrane degradation. This inflammatory microenvironment impairs the clearance of metabolic waste products, promoting the formation of drusen.

Cellular, Mitochondrial, and Lipid Homeostasis Alterations
Beyond classic complement activation, the CFH Y402H variant alters cellular metabolism and response to oxidative stress within the RPE. Studies using human induced pluripotent stem-cell-derived RPE (iPSC-RPE) show that cells harboring the high-risk (HR) CFH genotype exhibit significant baseline mitochondrial dysfunction. This is characterized by:
- Decreased mitochondrial mass and density.
- Reduced content of electron transport chain (ETC) proteins.
- Elevated levels of mitochondrial DNA (mtDNA) damage.
- Impaired bioenergetics, including lower ATP synthesis and reduced spare respiratory capacity under oxidative stress.

This bioenergetic deficit is exacerbated by environmental risk factors. For example, when iPSC-RPE cells are exposed to cigarette smoke extract (CSE)—the most consistent modifiable risk factor for AMD—cells with the high-risk CFH genotype show a dramatic drop in cell viability and an acceleration of mitochondrial decay compared to low-risk genotypes. This genetic-environmental synergy is clinically reflected in epidemiological data: smokers who are homozygous for the CFH high-risk CC genotype exhibit a 34-fold increased odds ratio for developing late-stage AMD compared to non-smokers carrying the low-risk TT genotype.
In vivo research using humanized mouse models has also revealed a novel connection between CFH and lipid metabolism. Aged transgenic mice expressing human CFH-H402 (homozygous risk) on a high-fat, cholesterol-enriched (HFC) diet developed classic AMD-like pathologies, including basal laminar deposits, multinucleated RPE cells, and rod-mediated visual dysfunction.
Intriguingly, biochemical analyses of these mice showed that these changes correlated with genotype-dependent alterations in systemic and eyecup lipoprotein levels—specifically a significant accumulation of Apolipoprotein B48 (ApoB48) and Apolipoprotein A1 (ApoA1) within the ocular tissues—rather than fluid-phase complement activation.This indicates that the CFH Y402H polymorphism plays a dual role in vivo, disrupting both complement regulation and lipid clearance at the RPE-Bruch's membrane interface.

The Chromosome 10q26 Locus: Deciphering the ARMS2-HTRA1 Controversy
The chromosome 10q26 locus is a major genetic contributor to AMD risk, accounting for approximately 30% of the genetic variance of the disease in European cohorts. This locus consists of a highly conserved ~5-kb linkage disequilibrium (LD) block containing three closely situated genes: PLEKHA1, ARMS2 (Age-Related Maculopathy Susceptibility 2), and HTRA1 (High-Temperature Requirement A Serine Peptidase 1).

The 10q26 risk variants carry a high odds ratio for AMD (ranging from 8.2 to 10.0) and show a strong association with the neovascular (wet) subtype and polypoidal choroidal vasculopathy (PCV). However, because of the high linkage disequilibrium across this region, identifying which specific gene is the primary causal driver of AMD pathogenesis has remained a subject of ongoing scientific debate.
│
HTRA1 Protease Overexpression (2-Fold)
│
Accelerated Cleavage of Fibulin 5, EFEMP1, & ApoE
│
Bruch's Membrane Fragmentation & Loss of Vascular SMA
│
Extracellular Matrix Disruption + Localized Lipid Pooling
│
Choroidal Neovascularization (nAMD / PCV)

The Causal Evidence and Pathological Role of HTRA1
The HTRA1 gene encodes a 51-kDa secreted serine protease that plays a key role in extracellular matrix (ECM) remodeling and turnover by degrading proteins like fibronectin, aggrecan, and various matrix components. The primary risk variant at this locus is the promoter polymorphism rs11200638 (G→A), which is associated with a twofold increase in HTRA1 transcription and secretion.
Several lines of evidence support HTRA1 as the primary causal factor at the 10q26 locus:
- eQTL and Expression Dynamics: Expression quantitative trait loci (eQTL) studies show that all AMD-associated variants that alter the expression of PLEKHA1 or ARMS2 also influence HTRA1 expression. In contrast, no variants altering only PLEKHA1 or ARMS2 expression are associated with AMD risk. Furthermore, HTRA1 is strongly expressed in human retinal tissue, particularly in the RPE and photoreceptors, whereas ARMS2 expression is extremely low to negligible in the adult retina.
- Extracellular Matrix Cleavage: Transgenic mice overexpressing HTRA1 exhibit structural fragmentation and loss of continuity in the elastic layer of Bruch's membrane. Overexpressed HTRA1 cleaves critical structural substrates, including fibulin 5, EFEMP1, and ApoE, reducing their levels by approximately 50%. This degradation leads to a loss of vascular smooth muscle actin (SMA) in the choroid, Bruch's membrane thickening, and loosening of collagen fibrils, which facilitates choroidal neovascularization.
- The R38X Nonsense Variant Dispute: A rare nonsense variant in ARMS2, rs2736911 (p.R38X), which results in a complete knock-out of ARMS2 protein translation, is not associated with AMD risk. If ARMS2 protein deficiency or dysfunction were the primary cause of AMD, this nonsense mutation would be expected to increase disease susceptibility. This finding casts doubt on the ARMS2-null causality hypothesis.

However, the role of HTRA1 is complex. Knockout mice lacking HTRA1 (Htra1 -/-) exhibit a progressive decline in rod and cone photoreceptor function, RPE atrophy, Bruch's membrane thickening, and the accumulation of sub-RPE deposits by 5 months of age. This indicates that normal, balanced levels of HTRA1 are critical for retinal homeostasis, and that either protease deficiency or excess can trigger macular pathology.

The Causal Evidence and Pathological Role of ARMS2
The ARMS2 gene encodes an 11.4-kDa primate-specific protein localized to the cytosol, mitochondria, and extracellular matrix. The primary risk polymorphism is a missense variant, rs10490924 (c.205G→T), which results in an alanine-to-serine substitution at position 69 (p.Ala69Ser or A69S).
Several mechanisms support a causal role for ARMS2 in AMD:
-
Mitophagy and Mitochondrial Power Failure: ARMS2 is concentrated in the mitochondria of photoreceptors and RPE cells, where it helps regulate mitophagy (the autophagic clearance of damaged, dysfunctional mitochondria). Impaired mitophagy is an early event in AMD pathogenesis. In early AMD, the levels of PINK1 (PTEN-induced kinase 1), a serine/threonine kinase that initiates mitophagy, are significantly decreased in perifoveal RPE cells. This deficiency leads to impaired mitophagy, the accumulation of damaged mitochondria, elevated ROS production, and a transition of RPE cells into a death-resistant, dysfunctional epithelial-mesenchymal transition (EMT) state. Mitophagy enhancers (e.g., spermidine, urolithin A) are being investigated as potential early-stage therapeutics to help restore the PINK1-Parkin pathway and protect RPE cells from oxidative stress.
-
Opsonization and Matrix Homeostasis: The ARMS2 protein binds directly to fibulin 6, a structural component of Bruch's membrane. The A69S variant impairs this binding, causing a loss of tissue elasticity, Bruch's membrane degradation, and anti-elastin autoimmunity. Additionally, ARMS2 is involved in the opsonization and complement-mediated clearance of apoptotic and necrotic cellular debris. A failure in this clearance pathway leads to the accumulation of lipid-rich deposits in Bruch's membrane, promoting drusen formation and vascular invasion.
-
CRISPR-Edited Functional Validation: CRISPR-mediated editing of the rs10490924 variant in iRPE cells has demonstrated that the presence of this high-risk allele is directly responsible for increased cellular ROS accumulation and a significant age-related decline in superoxide dismutase (SOD) expression and activity under oxidative stress. This validation confirms that the rs10490924 variant contributes to the high-oxidative-stress phenotype observed in patients with 10q26-associated AMD.

A summary of the evidence supporting both HTRA1 and ARMS2 as the causal driver at the 10q26 locus is detailed below:
| Feature / Metric | ARMS2 (p.A69S / rs10490924) | HTRA1 (rs11200638 Promoter Variant) |
| Gene Type |
Primate-specific intracellular/matrix protein. |
Highly conserved secreted serine protease. |
| Primary Site of Action |
Mitochondria (mitophagy) and extracellular matrix (fibulin 6 binding). |
Extracellular matrix (cleaves fibulin 5, EFEMP1, and ApoE). |
| Primary Cellular Impact |
Impairs mitophagy, reduces SOD activity, and increases ROS under stress. |
Degrades Bruch's membrane elasticity, thins choroidal vessels, and promotes CNV. |
| Clinical Association |
Strongly linked to wet AMD, geographic atrophy, and PCV. |
Strongly linked to wet AMD and Bruch's membrane fragmentation. |
| Supporting Causal Evidence |
CRISPR-edited iRPE cells show direct SOD decline and ROS elevation. |
eQTL studies show all AMD variants alter HTRA1; HTRA1 is highly expressed in adult retina. |
| Disputing / Complex Evidence |
The R38X nonsense mutation is not associated with AMD risk. |
Knockout (Htra1-/-) mice also develop progressive photoreceptor loss and RPE atrophy. |
Polygenic Risk Scores and Family History: Quantifying Cumulative Genetic Load
While major variants in CFH and ARMS2/HTRA1 are the strongest individual genetic risk factors for AMD, the clinical presentation is often influenced by the cumulative impact of many minor genetic variants. These minor variants are distributed across pathways involving lipid metabolism, extracellular matrix remodeling, and alternative complement activation. Clinical genomicists use Polygenic Risk Scores (PRS) to aggregate these small, additive genetic risks into a single predictive score.
Structure and Clinical Performance of Modern Polygenic Risk Scores
The clinical utility of PRS has improved with the integration of newly discovered genetic variants from large-scale GWAS. A comparative analysis of older models (such as PRS2016) and updated models (such as PRS2023) demonstrated a significant increase in predictive performance.
In a cohort of older individuals, a high PRS2023 (top quartile) was strongly associated with intermediate and late-stage AMD; over 27% of patients in this top quartile had advanced disease compared to less than 13% in the lowest quartile.
An integrated clinical model combining traditional, non-genetic risk factors (age, sex, smoking, and baseline lipid levels) with PRS2023 achieved an area under the receiver operating characteristic curve (AUC) of 91\% for predicting late-stage AMD, representing a 12% improvement over a clinical-only model (AUC} = 79%).
This predictive power is also being evaluated in prospective population screening trials, such as the Genetic Risk Assessment of Degenerative Eye Disease (GRADE) study, to identify high-risk individuals before they experience vision loss.
[Clinical-Only Prediction Model]
├── Predictors: Age, Sex, Smoking, Lipids
└── Accuracy: AUC = 79%
VS.
[Integrated Genomic Prediction Model]
├── Predictors: Clinical Factors + PRS2023
└── Accuracy: AUC = 91% (12% Absolute Gain)
Beyond prediction, polygenic risk stratification may help identify patients who are most likely to benefit from targeted therapeutic interventions. For example, in clinical trials of complement inhibitors, patients classified into the high complement-specific PRS group showed a drug effect size 1.6 to 2.3 times higher than the unstratified patient population. This suggests that complement-targeted therapies may be most effective in patients with a high baseline complement genetic load.

Integrating Minor Variants in Lipid and Extracellular Matrix Pathways
A comprehensive assessment of polygenic risk requires evaluating minor variants that regulate lipid transport, lipoprotein metabolism, and extracellular matrix remodeling at the RPE-Bruch's membrane interface. These minor variants, while carrying lower individual odds ratios than CFH or ARMS2, contribute to the cumulative risk profile.
| Gene | Locus | Primary Pathway | Biological Mechanism | Pathological Impact on Bruch's Membrane & Retina |
| ABCA1 | Chromosome 9 | Cholesterol Efflux & Reverse Lipid Transport |
Transports intracellular cholesterol and phospholipids to lipid-poor apolipoproteins, initiating nascent HDL synthesis. |
Impaired efflux leads to Tangier-like subretinal lipid accumulation, drusen growth, and inflammatory macrophage activation. |
| TIMP3 | Chromosome 22 | Extracellular Matrix (ECM) Remodeling |
Inhibits matrix metalloproteinases (MMPs) to prevent excessive ECM breakdown. |
Mutations compromise Bruch's membrane integrity, promoting drusen and mimicking Sorsby's fundus dystrophy. |
| LIPC | Chromosome 15 | Lipoprotein Metabolism & Hydrolysis |
Encodes hepatic lipase, which hydrolyzes triglycerides, HDLs, LDLs, and phospholipids. |
Dysregulation impairs lipoprotein clearance, driving early sub-RPE lipid-rich deposit deposition. |
| APOE | Chromosome 19q13.2 | Apolipoprotein / Lipid Transport |
Binds receptor complexes to facilitate systemic and local cholesterol uptake. |
Modulates the clearance of lipidaceous debris and RPE waste across Bruch's membrane. |
| CETP | Chromosome 16 | Cholesteryl Ester Transfer |
Mediates cholesteryl ester and triglyceride transfer between lipoprotein subfractions. |
Alters HDL metabolism and acts as a modifier of complement-driven drusen development. |
| LPL | Chromosome 8 | Lipoprotein Lipase Activity |
Catalyzes hydrolysis of triglycerides in chylomicrons and VLDLs. |
Modulates baseline retinal lipid clearance kinetics in conjunction with CFH. |
| C3(K155Q) | Chromosome 19 | Complement Activation |
Directly influences the rate of C3 activation and convertase stability. |
Accelerates systemic and local alternative pathway activation, raising odds for advanced disease. |
Composite Risk Modeling and Progression Kinetics
In clinical practice, integrating genetic risk with non-genetic variables allows for detailed risk stratification. A composite risk score combining 21 predictors—including 12 genetic variants (such as CFH, ARMS2/HTRA1, C3 K155Q, CFH R1210C, RAD51B, ABCA1, TIMP3, SLC16A8, and TGFBR1), family history, age, smoking status, and BMI—shows a strong association with the 5-year risk of progression to advanced AMD.
Comparing the 90th percentile of this composite risk score to the 10th percentile reveals a hazard ratio (HR) of 5.57for progression to advanced AMD, with predictive AUCs of 0.94 over 5 years and 0.92 over 12 years.
This composite modeling also reveals phenotypic differences:
- Having a first-degree family history of AMD (specifically greater or equal to 2 affected relatives) is more strongly associated with progression to neovascular wet AMD than to dry geographic atrophy.
- A high body mass index ({BMI equal or greater than 30 kg/m^2) consistently increases the risk of advanced AMD progression across all genomic backgrounds, with an independent hazard ratio of approximately 1.3.
- Nutritional supplementation (such as the AREDS2 protocol) is associated with a reduction in progression risk primarily for neovascular wet AMD, with little to no protective effect observed for geographic atrophy.
Protective Alleles: Innate Retinal Shields
Just as specific genetic variants accelerate retinal degeneration, the human genome also contains protective alleles that can buffer the eye against oxidative, metabolic, and inflammatory stress. These protective alleles often act by optimizing lipid clearance, stabilizing the extracellular matrix, or dampening unnecessary inflammatory cascades before they cause tissue damage.
PELI3 A307V and Innate Immune Modulation
A rare, non-synonymous protective variant, rs145732233, is located within the PELI3 gene on chromosome 11, resulting in an alanine-to-valine substitution at position 307 (A307V). Carrying this variant is associated with a highly significant reduction in advanced AMD risk (OR} = 0.14).
The PELI3 gene encodes the E3 ubiquitin ligase Pellino 3, a scaffold protein that helps transmit intracellular signals in response to Toll-like receptor (TLR) activation. TLRs, particularly TLR3, are expressed in RPE cells and recognize double-stranded RNA from pathogens or damaged host cells.
Under normal physiological conditions, over-activation of TLR3 can trigger a strong type I interferon response and promote pro-inflammatory cytokine release, contributing to chronic RPE degeneration.
The PELI3 A307V protective variant is thought to alter the signaling transduction cascade downstream of TLR3 :
- It enhances signaling through the interferon regulatory factor 7 (IRF7) pathway.
- This selective enhancement leads to a downregulation of type I interferon expression.
- Consequently, individuals carrying the A307V variant exhibit a milder, more regulated inflammatory response when exposed to cell debris or oxidative stress, protecting the surrounding RPE from inflammatory bystander damage.
The APOE ε4 vs ε2 Allelic Dichotomy
The apolipoprotein E (APOE) gene on chromosome 19q13.2 presents a striking genetic dichotomy, where different isoforms influence the risk of both Alzheimer's disease and AMD in opposite directions. The gene has three common polymorphic alleles: E2, E3 (the wild type), andE4.
│
┌─────────────────────────┴─────────────────────────┐
▼ ▼
[APOE ε4 Allele] [APOE ε2 Allele]
├── Risk HR = 0.66 (Highly Protective) ├── Risk HR = 1.15 (Risk-Conferring)
├── Increased Molecular Mobility ├── Enhanced Neovascularization
└── Facilitates Bruch's Membrane Waste Clearance └── Shifts Diagnosis 3.4 - 4.7 Years Earlier
The E4 allele (E3\E4 or E4\E4 genotypes) is highly protective against AMD, halving the risk of advanced geographic atrophy (OR = 0.35) and neovascular AMD (OR} = 0.58). The protective effect of the ApoE4 protein is thought to stem from its molecular mobility.
ApoE4 has a higher clearance rate and molecular mobility compared to other isoforms, which facilitates the transport and clearance of lipids, cholesterol, and RPE degradation products across Bruch's membrane. This rapid clearance prevents these waste products from accumulating within Bruch's membrane, reducing drusen formation and the subsequent risk of advanced AMD.
In contrast, the E2 allele (E3\E2 genotype) increases the risk of AMD (HR = 1.15) and shifts the mean age of clinical diagnosis earlier by up to 4.7\ years, particularly for the neovascular wet subtype. At the molecular level, ApoE2 has been shown to enhance abnormal vascularization and fibroblast activity, biological processes that can promote choroidal neovascularization.
Other Soluble Protective Variants
Additional protective alleles have been identified in alternative complement and metabolic pathway genes:
- CFH N1050Y (rs35274867): This rare, non-synonymous protective variant (OR = 0.76) is located within the CFHgene. It operates independently of other risk-conferring variants in the CFH gene, helping to limit complement pathway hyper-activation on self-tissues.
- CFB Protective Allele: Carriers of this variant exhibit lower circulating concentrations of Complement Factor B (CFB), a key catalytic component of the alternative pathway C3 convertase. Reducing systemic and local CFB levels directly decreases the rate of alternative pathway activation, protecting RPE cells from bystander damage.
- CTRB1 (rs8056814): A common protective variant located near the CTRB1 (Chymotrypsin B1) gene (OR = 0.71) is associated with a reduced risk of advanced AMD, suggesting a role for serine proteases in managing the baseline turnover of extracellular debris.
- CFHR1/CFHR3 Deletions: Homozygous deletions of CFHR1 and CFHR3 are highly protective against AMD.The CFHR1 and CFHR3 proteins can compete with CFH for binding to C3b. When these genes are deleted, this competition is eliminated, allowing CFH to regulate the alternative complement pathway more effectively.
Epigenetics and the Circulating Metabolome: Overwriting the Genetic Baseline
While a patient's inherited DNA sequence is fixed, the actual transcription and translation of these genes are modified by the metabolic environment through epigenetic mechanisms. Epigenetics, which includes DNA methylation, histone post-translational modifications, and chromatin remodeling, regulates which genes are actively expressed or silenced.
Understanding these mechanisms can help explain how lifestyle modifications interact with a patient's genetic profile to influence AMD progression.
Cigarette Smoke and Epigenetic Age Acceleration in the RPE
The primary environmental risk factor for AMD is cigarette smoking, which is associated with a 4-fold increase in the risk of advanced disease conversion. Research from Johns Hopkins Medicine (2026) using single-nuclear ATAC-sequencing (snATAC-seq) and single-nuclear RNA-sequencing (snRNA-seq) has helped define how cigarette smoke accelerates aging in RPE cells.
Analyzing RPE cells from young (3-month-old) and aged (12-month-old) mice exposed to cigarette smoke condensate (CSC) revealed that smoke exposure causes profound epigenetic changes :
- Formation of Dedifferentiated RPE Clusters: In both young and aged mice, acute CSC exposure led to the appearance of a transcriptomically distinct, dysfunctional RPE cell cluster. These cells exhibited a significant downregulation of core functional genes, such as Slc6a20a and Myrip, which are essential for normal RPE physiology.
- Decreased Chromatin Accessibility: CSC exposure significantly altered chromatin accessibility, reducing the physical accessibility of DNA regions containing genes needed for cellular repair and survival. This effectively "locks away" essential repair pathways.
•Loss of the "Safety Net" in Aged Cells: A key finding was the age-dependent difference in how cells responded to this stress. Young RPE cells exposed to CSC activated a transient protective response by upregulating a specific subset of "hallmarks of aging" genes involved in mitochondrial quality control, proteostasis (protein stability), autophagy, inflammation, and metabolic regulation. This activation allowed the young RPE cells to survive the acute stress. In contrast, aged RPE cells failed to activate these protective pathways under CSC stress, leading to chromatin closure, a failure of cellular repair, and cell death as measured by TUNEL labeling.
│
┌────────────────────────┴────────────────────────┐
▼ ▼
├── Epigenetic Stress ├── Epigenetic Stress
├── Active Epigenetic "Safety Net" ├── Failure to Activate "Safety Net"
├── Upregulated Mitophagy & Proteostasis ├── Chromatin Lock-Away of Repair Genes
└── Survival of Acute Exposure └── TUNEL-Positive Apoptotic Cell Death
Crucially, researchers identified 1,698 gene expression changes that were shared between these dysfunctional mouse models and human donor RPE tissues from smokers and early AMD patients. This suggests a highly conserved epigenetic mechanism through which environmental stress accelerates eye aging and cell death in patients with a predisposing genetic background.
The Circulating Metabolome as a Mediator of Genetic and Lifestyle Risks
The interaction between genetics, lifestyle, and disease is also reflected in the circulating blood metabolome. The EYE-RISK Project utilized a nuclear magnetic resonance (NMR) platform to analyze 146 plasma metabolites in 5,923 individuals, characterizing distinct metabolic signatures associated with different stages of AMD.
Early-Intermediate AMD Metabolomic Signature
In the early-to-intermediate stages of the disease, 61 plasma metabolites were significantly altered, with 94% of these belonging to lipid pathways. This stage is characterized by:
- Increased concentrations of high-density lipoprotein (HDL) subparticles, large HDLs, and Apolipoprotein A1 (OR = 1.2).
- Decreased levels of total cholesterol in very-low-density lipoproteins (VLDL), VLDL subparticles, triglycerides, and fatty acids (O} = 0.9).
- Reduced concentrations of non-lipid metabolites, including albumin, glycoprotein acetyls (related to inflammation), isoleucine, and citrate (OR = 0.9).
Late AMD Metabolomic Signature
As patients progress to advanced late-stage AMD, the metabolomic profile shifts from lipid alterations to markers of systemic amino acid depletion and ketone body elevation.
This stage is characterized by:
- A marked reduction in essential and non-essential amino acids, specifically tyrosine, histidine, leucine, phenylalanine, and valine (ORs:0.6 to 0.8).
- A significant elevation in the ketone bodies acetoacetate and 3-hydroxybutyrate (OR = 1.4), suggesting a metabolic shift toward fatty acid oxidation under conditions of energetic stress.
- A further reduction in citrate (OR = 0.8), which was the only metabolite consistently reduced across both early and late stages of AMD.
Mediation Analysis of the Metabolite Risk Score
By calculating a Metabolite Risk Score (MRS) based on these late-stage metabolites, researchers estimated the proportion of genetic and lifestyle risk mediated by circulating metabolites.
│
┌──────────────┴──────────────┐
▼ ▼
│ │
└──────────────┬──────────────┘
▼
Mediation analysis revealed that the MRS mediated 5.3% of the effect of the Genetic Risk Score (GRS) and 19.5% of the effect of the Lifestyle Risk Score (LRS) on late AMD progression. This indicates that while the genetic risk profile remains largely independent of circulating metabolites, nearly one-fifth of the risk associated with lifestyle choices (such as diet and smoking) is directly reflected in and mediated by the circulating metabolome.
A favorable lifestyle, characterized by high vegetable intake, was associated with higher circulating levels of protective amino acids and large HDLs, alongside lower levels of cholesterols, VLDL subclasses, and ketone bodies. Conversely, unfavorable lifestyles and smoking lowered citrate and increased inflammatory glycoprotein acetyls.
These findings suggest that targeted dietary and lifestyle interventions can help reshape the circulating metabolome, potentially mitigating some of the underlying genetic risk of advanced AMD progression.
A comparative summary of stage-specific systemic blood metabolites and their pathophysiological associations is detailed below:

Conclusions: Toward Personalized Ophthalmic Therapeutics
The translation of ophthalmic genetics into clinical practice is reshaping the management of age-related macular degeneration. AMD is no longer viewed as an inevitable consequence of aging, but rather as a highly heritable condition driven by specific molecular pathways. By identifying individual risk alleles, protective variants, and polygenic risk profiles, clinicians can transition from a standard, one-size-fits-all approach toward personalized clinical care.
This genetic framework has several key clinical implications:
- Targeted Complement Therapy: The clinical utility of complement inhibitors (such as C3 and Factor D blockers) is highly dependent on a patient's genetic profile. Stratifying patients based on a high complement pathway-specific PRS can identify those who are most likely to show a therapeutic response.
- Anti-HTRA1 Protease Inhibitors: For patients carrying the high-risk chromosome 10q26 locus where HTRA1 overexpression degrades Bruch's membrane, therapeutic strategies targeting HTRA1 peptidase activity are currently under development. This approach aims to preserve extracellular matrix integrity upstream of VEGF signaling, offering a complementary strategy to conventional anti-VEGF therapy for wet AMD.
- Mitigating Oxidative Stress: In patients with the ARMS2 rs10490924 variant, where mitochondrial dysfunction and attenuated SOD activity drive oxidative stress, targeted antioxidant therapies and chemical chaperones (such as sodium phenylbutyrate) represent promising avenues for preserving RPE cellular health.
- Epigenetic and Metabolomic Intervention: Because nearly 20% of lifestyle-associated risk is mediated through the circulating metabolome, targeted dietary interventions can be used to help counter genetic susceptibility.Remodeling the metabolome—by increasing protective amino acids and optimizing lipid profiles—can help preserve RPE function. Furthermore, identifying how environmental stressors like cigarette smoke cause chromatin changes in RPE cells may lead to the development of epigenetic therapies designed to restore chromatin accessibility at repair gene loci.
Genetic Variants and Markers Associated with Age-Related Macular Degeneration
|
Gene/Marker Name |
Locus |
Variant/Allele |
Biological Pathway |
Functional Impact |
Clinical Association |
Effect Size or Odds Ratio |
Source |
|
HTRA1 |
10q26 |
rs11200638 (Promoter G→A) |
ECM remodeling |
Increases HTRA1 transcription and secretion; degrades fibulin 5, EFEMP1, and ApoE. |
Risk (Strongly linked to wet AMD and Bruch's membrane fragmentation) |
OR 8.20 to 10.00 |
[1] |
|
ARMS2 |
10q26 |
rs10490924 (A69S) |
other |
Impairs mitophagy and mitochondrial power; increases ROS and decreases SOD activity. |
Risk (Strongly linked to wet AMD, geographic atrophy, and PCV) |
OR 8.20 to 10.00 |
[1] |
|
CFH |
1q23-32 |
rs1061170 (Y402H) |
complement |
Alters electrostatic charge of SCR7 binding pocket; reduces binding to polyanions and Bruch's membrane. |
Risk (5- to 7-fold increase for CC genotype) |
OR 2.20 to 7.00 |
[1] |
|
APOE ε4 |
19q13.2 |
ε4 allele |
lipid metabolism |
Higher clearance rate and molecular mobility of lipids across Bruch's membrane. |
Protective against GA and neovascular AMD |
OR 0.35 to 0.58 |
[1] |
|
PELI3 |
Chromosome 11 |
rs145732233 (A307V) |
other |
Enhances IRF7 pathway and downregulates type I interferon response. |
Protective against advanced AMD |
OR 0.14 |
[1] |
|
APOE ε2 |
19q13.2 |
ε2 allele |
lipid metabolism |
Enhances abnormal vascularization and fibroblast activity. |
Risk (Earlier diagnosis and neovascular wet subtype) |
HR 1.15 |
[1] |
|
Acetoacetate and 3-hydroxybutyrate |
Systemic (Blood) |
Ketone bodies |
other |
Indicates systemic ketogenesis and metabolic shift under energetic strain. |
Risk/Association (Late AMD) |
OR 1.40 |
[1] |
|
Citrate |
Systemic (Blood) |
Metabolite |
other |
Points to mitochondrial TCA cycle disruption across all stages. |
Risk/Association (Decreased in early and late AMD) |
OR 0.80 to 0.90 |
[1] |
|
Tyrosine, Histidine, Leucine, Phenylalanine, Valine |
Systemic (Blood) |
Amino acid group |
other |
Systemic amino acid depletion indicating impaired tissue repair capacity. |
Risk/Association (Late AMD) |
OR 0.60 to 0.80 |
[1] |
|
C3 (K155Q) |
Chromosome 19 |
K155Q |
complement |
Influences rate of C3 activation and convertase stability. |
Risk (Advanced disease) |
Not in source |
[1] |
|
ABCA1 |
Chromosome 9 |
Not in source |
lipid metabolism |
Impaired cholesterol efflux leads to subretinal lipid accumulation. |
Risk (Drusen growth and inflammatory macrophage activation) |
Not in source |
[1] |
|
TIMP3 |
Chromosome 22 |
Not in source |
ECM remodeling |
Compromises Bruch's membrane integrity by failing to inhibit MMPs. |
Risk (Drusen and mimics Sorsby's fundus dystrophy) |
Not in source |
[1] |
Genetic & Hereditary Risk in Age-Related Macular Degeneration (AMD)
In summary, mapping the genetic and epigenetic landscape of AMD allows clinicians to identify the specific molecular pathways driving a patient's disease. This genetic stratification is the foundation of precision medicine in ophthalmology, enabling targeted interventions designed to preserve visual function and support long-term ocular health.
Frequently Asked Questions (FAQs):
What is complement-specific Risk factor?
A complement-specific risk factor refers to a genetic variation within the alternative complement pathway—a self-amplifying part of the innate immune system—that predisposes an individual to Age-Related Macular Degeneration (AMD) by driving localized inflammation and tissue damage. While the complement system normally neutralizes pathogens and clears cellular debris, specific genetic mutations cause it to become overactive and attack healthy host tissues.
The most powerful complement-specific risk factor is the Y402H variant in the Complement Factor H (CFH) gene. Under normal conditions, the CFH protein circulates in the blood and acts as the primary inhibitor of the alternative complement pathway, protecting healthy retinal tissues. However, the Y402H mutation reduces the protein's ability to anchor to crucial structures like Bruch's membrane and the surface of the retinal pigment epithelium (RPE). When this regulatory control is lost, the complement cascade forms unchecked, leading to localized cell lysis, degradation of Bruch's membrane, and the accumulation of metabolic waste known as drusen. Carrying two copies of this high-risk CFH variant elevates a patient's relative risk of AMD by 5- to 7-fold.
Another notable complement-specific risk factor is the K155Q variant in the C3 gene. This mutation directly accelerates both local and systemic activation of the alternative pathway, significantly raising the odds of developing advanced disease.
To measure a patient's overall vulnerability to this specific disease mechanism, clinicians can aggregate these individual mutations into a complement-specific Polygenic Risk Score (PRS), which quantifies their cumulative complement genetic load. Stratifying patients based on these complement-specific risk factors is a critical step in personalized ophthalmic medicine. For instance, clinical trials indicate that patients with a high complement-specific PRS respond much better to targeted complement therapies (such as C3 and Factor D blockers), demonstrating a drug effect size 1.6 to 2.3 times higher than the general, unstratified patient population.
What specific genes are linked to the complement-specific PRS?
The provided sources do not explicitly list all the specific genes that make up the complement-specific Polygenic Risk Score (PRS). However, they do identify several major genes within the alternative complement pathway that drive a patient's complement-specific genetic load:
- CFH (Complement Factor H): Identified as one of the strongest susceptibility loci for AMD, this gene includes major high-risk variants like Y402H and R1210C, as well as the protective N1050Y variant.
- C3: The K155Q variant in this gene directly accelerates systemic and local activation of the alternative complement pathway.
- CFB (Complement Factor B): Protective variants in this gene help decrease the rate of alternative pathway activation by lowering circulating CFB levels.
- CFHR1 and CFHR3: Homozygous deletions in these genes are highly protective, as they stop the CFHR proteins from competing with CFH, allowing CFH to more effectively regulate the complement pathway.
While an exact list for the isolated complement-specific PRS is absent, the sources mention that clinicians use a broader composite risk score to predict advanced AMD progression. This comprehensive score incorporates the complement genes CFH and C3, alongside 10 other genetic variants, including ARMS2/HTRA1, RAD51B, ABCA1, TIMP3, SLC16A8, and TGFBR1.
Are there other genes in the PRS besides complement ones?
Yes, comprehensive Polygenic Risk Scores (PRS) and composite risk models for Age-Related Macular Degeneration (AMD) incorporate many genes outside of the complement system. While major complement variants carry high individual risk, the clinical presentation of AMD is heavily influenced by the cumulative impact of minor genetic variants distributed across other biological pathways, particularly those governing lipid metabolism and extracellular matrix (ECM) remodeling.
Specific non-complement genes included in these comprehensive risk scores include:
- Lipid Transport and Metabolism Genes: Variants in ABCA1, LIPC, APOE, CETP, and LPL are factored into the cumulative risk profile because they alter how lipids, triglycerides, and cholesterol are processed and cleared in the eye. For instance, impaired ABCA1 function leads to subretinal lipid accumulation and drusen growth, while APOE modulates the clearance of lipidaceous waste across Bruch's membrane.
- Extracellular Matrix Remodeling Genes: The TIMP3 gene is included because it inhibits matrix metalloproteinases to prevent excessive ECM breakdown; mutations here compromise the structural integrity of Bruch's membrane.
- The Chromosome 10q26 Locus: The highly significant ARMS2 and HTRA1 genes are core non-complement components of genetic risk models. These genes drive pathology through distinctly different pathways: ARMS2 is linked to mitochondrial mitophagy and elevated reactive oxygen species (ROS), while HTRA1 is a secreted protease that cleaves structural matrix components, promoting choroidal neovascularization.
- Other Associated Genes: Composite risk models also integrate genetic variants in RAD51B, SLC16A8, and TGFBR1.
By aggregating these small, additive genetic risks from non-complement pathways alongside major complement drivers (such as CFH and C3), clinical genomicists can calculate a highly predictive composite PRS. When this comprehensive genetic load is combined with non-genetic variables like age, BMI, and smoking status, it achieves an impressive 91% to 94% accuracy in predicting a patient's risk of progressing to late-stage AMD over time.
What do the ARMS2 and HTRA1 genes do?
The ARMS2 (Age-Related Maculopathy Susceptibility 2) and HTRA1 (High-Temperature Requirement A Serine Peptidase 1) genes are situated close together in a highly conserved block on the chromosome 10q26 locus. This locus is a major genetic contributor to AMD, accounting for roughly 30% of the disease's genetic variance in European populations.
Because these two genes are in high "linkage disequilibrium" (meaning they are inherited together very frequently), there has been an ongoing scientific debate over which gene is the primary driver of AMD pathogenesis. Current evidence indicates that they both play distinct but highly significant roles in retinal health and disease:
The Role of the HTRA1 Gene The HTRA1 gene encodes a secreted serine protease, an enzyme that operates in the extracellular matrix (ECM).
- Normal Function: Its primary role is to manage ECM remodeling and turnover by breaking down proteins such as fibronectin, aggrecan, and various other matrix components. Normal, balanced levels of this protein are essential for maintaining retinal homeostasis; in fact, animal models completely lacking HTRA1 experience progressive photoreceptor loss and retinal atrophy.
- Role in AMD: The primary genetic risk variant for HTRA1 causes a twofold increase in the transcription and secretion of this enzyme. This overexpression leads to excessive cleavage of critical structural substrates in the eye, including fibulin 5 and ApoE. As a result, the elastic layer of Bruch's membrane fragments, collagen fibrils loosen, and blood vessels thin out, which strongly facilitates the abnormal blood vessel growth (choroidal neovascularization) characteristic of wet AMD.
The Role of the ARMS2 Gene Unlike HTRA1, ARMS2 encodes a primate-specific protein that functions within the cell—specifically inside the cytosol and mitochondria—as well as in the extracellular matrix.
- Normal Function: Within the mitochondria of retinal pigment epithelium (RPE) cells and photoreceptors, ARMS2 helps regulate mitophagy, which is the vital process of clearing away damaged and dysfunctional mitochondria. Outside the cell, ARMS2 binds directly to a structural component called fibulin 6 to maintain the elasticity of Bruch's membrane, and it helps flag dead cellular debris for clearance.
- Role in AMD: The primary high-risk variant of ARMS2 (A69S) disrupts these protective functions. It impairs mitophagy, causing damaged mitochondria to pile up and produce dangerous levels of reactive oxygen species (ROS). It also reduces the activity of superoxide dismutase (SOD), an important antioxidant defense. Furthermore, the mutant protein fails to properly bind to fibulin 6 and fails to clear necrotic debris, leading to a loss of tissue elasticity and the accumulation of lipid-rich deposits (drusen) that invite vascular invasion.
Ultimately, both genes drive AMD pathology through distinctly different pathways: HTRA1 primarily damages the extracellular structure through excessive enzymatic degradation, while ARMS2 drives cellular power failure (mitochondrial dysfunction), oxidative stress, and impaired waste clearance.
What does the HTRA1 overexpression do to Bruch's membrane?
Overexpression of the HTRA1 gene, which encodes an enzyme responsible for extracellular matrix remodeling, leads to the excessive breakdown of critical structural components in the eye. When overexpressed, the HTRA1 enzyme abnormally cleaves and degrades key substrates like fibulin 5, EFEMP1, and ApoE, reducing their levels by roughly 50%.
This aggressive degradation has several destructive effects on Bruch's membrane:
- Structural Fragmentation: It causes the elastic layer of Bruch's membrane to fragment, resulting in a loss of continuity and elasticity.
- Thickening and Loosening: The abnormal cleavage leads to an overall thickening of Bruch's membrane and the loosening of its collagen fibrils.
Ultimately, this structural compromise of Bruch's membrane—coupled with the loss of vascular smooth muscle actin in the choroid—thins choroidal blood vessels and paves the way for choroidal neovascularization (the abnormal blood vessel growth characteristic of wet AMD).
How do anti-HTRA1 protease inhibitors help treat wet AMD?
Building on our previous discussion about how the overexpressed HTRA1 enzyme aggressively degrades Bruch's membrane, anti-HTRA1 protease inhibitors are designed to stop this specific structural breakdown before it leads to wet AMD.
Here is how these inhibitors work as a treatment:
- Targeting the Root Enzymatic Cause: In patients who carry the high-risk chromosome 10q26 genetic variants, the HTRA1 enzyme is overproduced and excessively breaks down the extracellular matrix. Anti-HTRA1 inhibitors specifically target and neutralize this overactive peptidase activity.
- Preserving Structural Integrity: By blocking the excess enzyme, these inhibitors prevent the fragmentation and loosening of Bruch's membrane, preserving its physical integrity.
- Acting "Upstream" of VEGF: The hallmark of wet AMD is abnormal blood vessel growth, which is driven by VEGF signaling. Because the structural breakdown of Bruch's membrane paves the way for these vessels to invade, anti-HTRA1 inhibitors act upstream of VEGF signaling. They aim to prevent the structural collapse that triggers the pathological vessel growth in the first place.
Ultimately, clinical researchers view anti-HTRA1 protease inhibitors as a complementary strategy to conventional anti-VEGF therapy. While standard anti-VEGF injections manage the abnormal blood vessels once they appear, anti-HTRA1 therapies aim to protect the foundational structure of the eye from being degraded by the patient's own genetics.
What other biological pathways does the 10q26 locus affect?
The chromosome 10q26 locus—which consists of a tightly linked block containing the ARMS2, HTRA1, and PLEKHA1genes—influences a diverse array of biological pathways that dictate retinal health and disease. Through the combined actions of these genes, this locus directly impacts the following cellular and metabolic processes:
- Extracellular Matrix (ECM) Remodeling and Homeostasis: This locus heavily regulates the physical structure of the eye. HTRA1 drives ECM turnover by secreting serine proteases that degrade structural proteins like fibronectin, aggrecan, fibulin 5, EFEMP1, and ApoE. At the same time, ARMS2 normally supports matrix homeostasis by binding to fibulin 6 to maintain the elasticity of Bruch's membrane.
- Mitochondrial Quality Control (Mitophagy): ARMS2 is concentrated within the mitochondria of photoreceptors and RPE cells, where it regulates mitophagy—the crucial autophagic clearance of damaged and dysfunctional mitochondria. When this pathway is impaired, it leads to early pathogenic events and cellular power failure.
- Oxidative Stress Regulation: The locus directly influences the cell's ability to manage oxidative stress. Genetic variants here cause a significant, age-related decline in the protective antioxidant enzyme superoxide dismutase (SOD) and lead to the dangerous accumulation of reactive oxygen species (ROS).
- Epithelial-Mesenchymal Transition (EMT): When the PINK1-initiated mitophagy pathway (associated with ARMS2) fails, it triggers retinal pigment epithelium (RPE) cells to undergo a harmful transition. The cells enter a dysfunctional, death-resistant state known as an epithelial-mesenchymal transition (EMT).
- Opsonization and Cellular Waste Clearance: The ARMS2 protein functions in the opsonization pathway, which coordinates the complement-mediated tagging and clearance of apoptotic and necrotic cellular debris. When this pathway is disrupted, it leads to the accumulation of lipid-rich deposits (drusen) in Bruch's membrane, which subsequently promotes abnormal vascular invasion.
How do genetic risk scores predict 5-year progression risk?
Genetic risk scores predict the 5-year risk of progression to advanced Age-Related Macular Degeneration (AMD) by integrating a patient's genetic profile with traditional clinical and lifestyle factors to create a highly accurate composite risk score.
To calculate this 5-year progression risk, clinical genomicists use a model consisting of 21 predictors. This includes 12 specific genetic variants—incorporating major drivers like CFH, ARMS2, and HTRA1, as well as minor variants across lipid and extracellular matrix pathways like ABCA1, TIMP3, and C3. These genetic factors are then combined with non-genetic variables, including the patient's age, family history, Body Mass Index (BMI), and smoking status.
By evaluating these combined factors, the composite risk score achieves an impressive predictive accuracy for advanced AMD, demonstrating an Area Under the Curve (AUC) of 0.94 over a 5-year period (and 0.92 over a 12-year period). When comparing patients in the 90th percentile of this risk score to those in the 10th percentile, those with the highest scores face a hazard ratio of 5.57 for progressing to advanced disease.
Furthermore, this comprehensive 5-year progression modeling reveals several specific clinical insights regarding how non-genetic factors influence the trajectory of the disease:
- Family History: Having two or more affected first-degree relatives is more strongly associated with progressing to neovascular (wet) AMD than to dry geographic atrophy.
- Obesity: A BMI of 30 or higher consistently increases the risk of progression to advanced AMD (with a hazard ratio of approximately 1.3) regardless of the patient's underlying genomic background.
- Supplementation: Adherence to nutritional protocols, such as AREDS2, successfully reduces the 5-year progression risk primarily for wet AMD, but shows little to no protective effect against dry geographic atrophy.
What does a high BMI do to AMD progression?
A high body mass index (BMI of 30 kg/m² or higher) acts as a consistent risk factor that increases a patient's likelihood of progressing to advanced Age-Related Macular Degeneration (AMD).
According to clinical progression models, obesity carries an independent hazard ratio of approximately 1.3 for driving the disease to its late stages. Crucially, this heightened risk is observed across all genomic backgrounds, meaning that a high BMI exacerbates AMD progression regardless of the specific genetic vulnerabilities a patient might carry.
Because of its significant and independent impact on disease trajectory, a patient's BMI is integrated alongside genetics, age, and smoking status as a core component of the 21-predictor composite risk score used by clinicians to forecast a patient's 5-year risk of developing advanced AMD.
What makes the CFH and C3 variants so high-risk?
The CFH (Complement Factor H) and C3 genetic variants are considered high-risk for Age-Related Macular Degeneration (AMD) because they fundamentally dysregulate the alternative complement pathway—a self-amplifying part of the innate immune system. Normally, this pathway safely clears cellular debris and neutralizes pathogens, but these specific mutations cause the immune system to over-activate and aggressively attack healthy retinal tissues.
Here is what makes each variant specifically high-risk:
The CFH Y402H Variant (Loss of Regulatory Control) The CFH protein normally circulates in the blood and acts as the primary "brake" for the alternative complement pathway, preventing it from damaging host tissues.
- Anchoring Failure: The high-risk Y402H variant alters the electrostatic charge of the protein's binding pocket (the SCR7 domain). Because of this change, the mutant CFH protein loses its affinity for protective ligands and fails to physically anchor properly to Bruch's membrane and the surface of the retinal pigment epithelium (RPE).
- Unchecked Immune Attack: Without CFH firmly anchored to these tissues to suppress the immune response, the alternative pathway cascade forms unchecked. This triggers the downstream assembly of the membrane attack complex (MAC), which induces localized cell lysis, the degradation of Bruch's membrane, and impairs the clearance of metabolic waste, leading to the rapid build-up of drusen.
- Mitochondrial and Lipid Disruption: As we noted in our earlier discussion regarding smoking and diet, the CFHhigh-risk variant also drives severe baseline mitochondrial dysfunction and actively disrupts lipid clearance at the RPE-Bruch's membrane interface.
The C3 K155Q Variant (Accelerated Activation) While the CFH variant removes the "brakes" of the complement pathway, the high-risk C3 variant effectively steps on the "gas."
- Increased Activation: The K155Q variant in the C3 gene directly influences the rate of C3 activation and increases the stability of the convertase enzyme that drives the immune cascade.
- Accelerated Damage: This mutation rapidly accelerates both systemic and local activation of the alternative complement pathway, significantly elevating a patient's risk for advanced disease.
Ultimately, these two variants are highly destructive because they compromise the same biological pathway—one utterly fails to protect the tissue from inflammation, and the other actively accelerates the inflammatory immune attack.
Are there other genes in the PRS besides complement ones?
Yes, as we touched on earlier, comprehensive Polygenic Risk Scores (PRS) and composite risk models for Age-Related Macular Degeneration (AMD) incorporate a wide variety of genes outside of the complement system. While major complement variants carry high individual risk, a comprehensive PRS integrates minor variants that are primarily distributed across lipid metabolism and extracellular matrix (ECM) remodeling pathways.
Specific non-complement genes included in these risk models include:
- The Chromosome 10q26 Locus (ARMS2 and HTRA1): As we explored in our recent discussions, these are major non-complement contributors to AMD risk. They drive pathology through distinctly different pathways: HTRA1aggressively degrades the ECM and Bruch's membrane, while ARMS2 drives mitochondrial dysfunction and oxidative stress.
- Lipid Transport and Metabolism Genes: Variants in ABCA1, LIPC, APOE, CETP, and LPL are integrated into the cumulative risk profile because they manage how lipids, triglycerides, and cholesterol are processed and cleared in the eye.
- Impaired ABCA1 function disrupts reverse lipid transport, leading to subretinal lipid accumulation and drusen growth.
- APOE modulates the clearance of lipidaceous waste across Bruch's membrane; notably, the $\varepsilon4$ allele acts as a powerful protective shield against AMD, while the $\varepsilon2$ allele increases risk.
- Dysregulation in LIPC impairs lipoprotein clearance, driving early lipid deposits.
- Extracellular Matrix Remodeling Genes: The TIMP3 gene is included because its normal function is to inhibit matrix metalloproteinases to prevent excessive ECM breakdown. Mutations in this gene compromise the structural integrity of Bruch's membrane.
- Immune Modulation Genes: A patient's score may be influenced by protective alleles in genes like PELI3, which alters intracellular signaling to prevent chronic, damaging inflammation in the retinal pigment epithelium.
- Additional Predictive Genes: The 21-predictor composite risk score used to forecast 5-year disease progression incorporates 12 specific genetic variants, which also include RAD51B, SLC16A8, and TGFBR1 alongside the complement drivers.
By aggregating these small, additive genetic risks from non-complement pathways alongside major complement drivers, clinical genomicists can quantify a patient's cumulative genetic load. When combined with lifestyle factors like BMI and smoking, this yields a highly accurate predictive model for anticipating advanced AMD.
How do C3 and Factor D blockers help high-risk patients?
C3 and Factor D blockers are targeted complement inhibitors designed to counteract the over-activation of the alternative complement pathway. These therapies are particularly beneficial for high-risk patients who carry a high baseline complement genetic load, a vulnerability that clinicians measure using a high complement pathway-specific Polygenic Risk Score (PRS).
As discussed previously, in patients with high-risk genetic variants (such as the CFH Y402H loss-of-function or C3 K155Q accelerated-activation mutations), the innate immune system forms an unchecked cascade that aggressively attacks healthy retinal tissues. Because C3 and Factor D blockers specifically target the very molecular pathway driving the disease in these individuals, these patients are significantly more likely to show a strong therapeutic response.
Clinical trials have demonstrated the power of this targeted approach: when patients are stratified and treated based on having a high complement-specific PRS, they exhibit a drug effect size 1.6 to 2.3 times higher than the general, unstratified patient population. Ultimately, utilizing C3 and Factor D blockers in this manner represents a major step toward personalized ophthalmic therapeutics, allowing clinicians to match specific medical interventions directly to a patient's underlying genetic vulnerabilities
What is the role of the K155Q variant in the C3 gene?
The K155Q variant in the C3 gene, located on chromosome 19, acts as a high-risk genetic driver for Age-Related Macular Degeneration (AMD) by aggressively over-activating the alternative complement pathway.
Specifically, the role of this variant includes:
- Accelerated Enzyme Activation: The K155Q mutation directly increases the rate of C3 activation and enhances the stability of the convertase enzyme.
- Increased Tissue Damage: By stabilizing this enzyme, the variant accelerates both local and systemic activation of the alternative complement cascade. This rapid activation cleaves the C3 protein into C3a and C3b, which drives the downstream assembly of the membrane attack complex (MAC). The resulting accumulation of MAC induces localized cell death (lysis), degrades Bruch's membrane, and impairs metabolic waste clearance to promote drusen formation.
- Risk Prediction: Because it significantly raises the odds for advanced disease, the C3 K155Q mutation is incorporated as a core component of the 21-predictor composite risk score. Clinicians use this specific variant, alongside others like CFH and ARMS2/HTRA1, to accurately model a patient's 5-year hazard ratio for progressing to late-stage AMD.
- Ultimately, while variants like CFH Y402H remove the "brakes" by failing to inhibit the complement system, the C3 K155Q variant effectively steps on the "gas" to speed up this destructive inflammatory cascade.
What is the role of the APOE gene in AMD?
The APOE (Apolipoprotein E) gene, located on chromosome 19q13.2, plays a fundamental role in lipid transport, specifically by modulating how cholesterol, lipidaceous debris, and cellular waste from the retinal pigment epithelium (RPE) are cleared across Bruch's membrane.
What makes the APOE gene particularly unique in the context of Age-Related Macular Degeneration (AMD) is its striking genetic dichotomy—meaning different versions (alleles) of the gene influence the disease in completely opposite directions.
The gene has three common polymorphic alleles: ε2, ε3 (the standard wild type), and ε4. The roles of the two variant alleles in AMD are as follows:
- The Protective ε4 Allele: As touched on in our earlier discussions about the composite risk score, carrying the ε4 allele acts as a powerful shield against AMD. It essentially halves a patient's risk of developing advanced geographic atrophy and neovascular (wet) AMD. This protective effect occurs because the ApoE4 protein has higher molecular mobility and a faster clearance rate than the other isoforms. This rapid clearance efficiently transports lipids and waste across Bruch's membrane, preventing the accumulation of debris that forms drusen and avoiding the progression of advanced disease.
- The High-Risk ε2 Allele: In stark contrast, the ε2 allele actively increases a patient's risk of AMD and can shift the onset of clinical diagnosis earlier by up to 4.7 years, particularly for the neovascular wet subtype. At a molecular level, the ApoE2 protein is shown to enhance fibroblast activity and abnormal vascularization. These biological processes directly promote choroidal neovascularization, which is the destructive, abnormal blood vessel growth characteristic of wet AMD
How do lipid metabolism genes affect Bruch's membrane?
Lipid metabolism genes play a crucial role in maintaining the health of Bruch's membrane by regulating how cholesterol, triglycerides, and metabolic waste are transported and cleared from the eye. When these genes are mutated or dysregulated, they cause lipid-rich deposits (drusen) to build up, structurally compromising the interface between the retinal pigment epithelium (RPE) and Bruch's membrane.
Based on the sources, here is how specific lipid metabolism genes affect Bruch's membrane:
- ABCA1 (Cholesterol Efflux): This gene normally transports intracellular cholesterol and phospholipids to help synthesize high-density lipoproteins (HDL). When ABCA1 function is impaired, it disrupts reverse lipid transport, leading to subretinal lipid accumulation, the growth of drusen along Bruch's membrane, and the activation of inflammatory macrophages.
- APOE (Apolipoprotein Transport): This gene modulates the clearance of lipidaceous debris and RPE waste across Bruch's membrane. As we discussed previously, the highly protective ε4 allele facilitates rapid transport and clearance of lipids, successfully preventing waste products from accumulating within Bruch's membrane. Conversely, the high-risk ε2 allele fails to clear this debris as effectively and actively enhances fibroblast activity and abnormal vascularization.
- LIPC (Lipoprotein Hydrolysis): This gene encodes an enzyme that hydrolyzes triglycerides, HDLs, and LDLs. Dysregulation of LIPC impairs the clearance of lipoproteins, directly driving the early deposition of lipid-rich sub-RPE deposits onto Bruch's membrane.
- CETP and LPL: CETP alters HDL metabolism and modifies how complement-driven drusen develop, while LPL modulates the baseline kinetics of retinal lipid clearance.
- Furthermore, genes primarily known for other functions can also severely impact lipid clearance at this barrier. For example, the high-risk Y402H variant of the CFH gene not only triggers an unchecked immune attack but also actively disrupts lipid clearance at the RPE-Bruch's membrane interface. In animal models, this high-risk variant combined with a high-fat diet caused a significant accumulation of Apolipoprotein B48 (ApoB48) and Apolipoprotein A1 (ApoA1) in ocular tissues, driving the formation of classic AMD-like basal laminar deposits.
Ultimately, these minor and major genetic variants dictate how efficiently the eye processes and removes its structural waste; when these clearance mechanisms fail, the resulting lipid pile-up on Bruch's membrane creates the foundational pathology of macular degeneration.
What is the relationship between the ARMS2 gene and mitophagy?
The ARMS2 (Age-Related Maculopathy Susceptibility 2) gene plays a critical regulatory role in mitophagy, which is the essential cellular process responsible for clearing away damaged and dysfunctional mitochondria.
Here is how the ARMS2 gene and mitophagy interact to influence retinal health:
- Normal Regulation: The ARMS2 protein is highly concentrated within the mitochondria of retinal pigment epithelium (RPE) cells and photoreceptors. Under healthy conditions, it helps regulate the necessary autophagic clearance of aging mitochondria to maintain cellular energy production and prevent toxicity.
- Pathological Impairment: A breakdown in mitophagy is identified as an early event in the pathogenesis of Age-Related Macular Degeneration (AMD). In early AMD, the levels of PINK1—a specific kinase that initiates the mitophagy process—are significantly decreased in RPE cells. The primary high-risk ARMS2 genetic variant (A69S or rs10490924) directly impairs this vital clearance mechanism.
- Cellular Consequences: Because the mutant ARMS2 protein fails to execute proper mitophagy, damaged and dysfunctional mitochondria pile up inside the cells. This accumulation leads to a severe state of oxidative stress, characterized by elevated reactive oxygen species (ROS) and a significant decline in protective antioxidants like superoxide dismutase (SOD).
- Epithelial-Mesenchymal Transition: Ultimately, the failure of the PINK1-initiated mitophagy pathway and the resulting mitochondrial power failure force the RPE cells into a dysfunctional, death-resistant state known as an epithelial-mesenchymal transition (EMT).
- To counteract this mechanism, researchers are currently investigating mitophagy enhancers (such as spermidine and urolithin A) as potential early-stage therapeutics to help restore the PINK1 pathway and protect RPE cells from oxidative stress
How ApoE4 protects against geographic atrophy?
The ApoE4 protein, which is encoded by the protective ε4 allele of the APOE gene, protects against advanced geographic atrophy primarily through its superior molecular mobility and higher clearance rate compared to other isoforms.
Because of this heightened mobility, ApoE4 highly efficiently facilitates the transport and removal of lipids, cholesterol, and retinal pigment epithelium (RPE) degradation products across Bruch's membrane. By rapidly sweeping this metabolic waste away, the ApoE4 protein prevents dangerous lipidaceous debris from accumulating at the crucial interface between the RPE and Bruch's membrane.
Ultimately, because this debris is successfully cleared rather than left to pile up, it significantly reduces the formation of drusen (lipid-rich deposits), thereby protecting the structural integrity of the retina and cutting a patient's risk of developing advanced geographic atrophy by nearly half.
How ARMS2 mutations cause mitochondrial power failure?
The high-risk ARMS2 mutation (specifically the missense variant rs10490924 or A69S) causes mitochondrial power failure by fundamentally disrupting mitophagy, the cellular process responsible for clearing away damaged mitochondria.
Normally, the ARMS2 protein is concentrated within the mitochondria of retinal pigment epithelium (RPE) cells and photoreceptors, where it helps regulate this vital autophagic clearance. However, the mutated ARMS2 protein directly impairs this process. In the presence of this mutation, levels of PINK1 (PTEN-induced kinase 1)—an enzyme that is essential for initiating mitophagy—are significantly decreased in RPE cells.
Because this clearance mechanism is compromised, damaged and dysfunctional mitochondria pile up inside the cells. This accumulation of faulty mitochondria creates a state of severe bioenergetic failure and oxidative stress, characterized by:
- Elevated Reactive Oxygen Species (ROS): The piled-up, damaged mitochondria continuously produce dangerous levels of ROS.
- Loss of Antioxidant Defenses: CRISPR-edited functional validation shows that the presence of the high-risk ARMS2 allele directly causes a significant, age-related decline in the expression and activity of superoxide dismutase (SOD), a crucial protective antioxidant enzyme.
Ultimately, this unmanageable oxidative stress and mitochondrial power failure force the RPE cells to undergo an epithelial-mesenchymal transition (EMT), entering a dysfunctional, death-resistant state that serves as an early step in macular degeneration
How do C3 blockers improve treatment for high-risk patients?
C3 blockers improve treatment for high-risk patients by specifically targeting the biological pathway that drives their disease, making the therapy significantly more effective for them than for the general population.
Patients who are considered high-risk in this context carry a high baseline complement genetic load, which clinicians measure using a high complement pathway-specific Polygenic Risk Score (PRS). Because their specific genetic makeup (involving variants like CFH and C3) causes the alternative complement pathway to over-activate and attack healthy tissue, these patients are uniquely positioned to benefit from drugs that block this exact process.
When clinicians use genetic stratification to identify these high-risk patients, clinical trials show that C3 blockers (and other complement inhibitors like Factor D blockers) yield a drug effect size 1.6 to 2.3 times higher than they do in the general, unstratified patient population. Ultimately, utilizing C3 blockers in this targeted manner represents a major advancement in personalized ophthalmic therapeutics, allowing doctors to match specific medications directly to the patient's underlying molecular vulnerabilities
Which factors are included in the 21-predictor risk model?
The 21-predictor composite risk model integrates both genetic and non-genetic variables to accurately forecast a patient's 5-year risk of progressing to advanced Age-Related Macular Degeneration (AMD).
Specifically, the model incorporates the following factors:
- 12 Genetic Variants: The score calculates cumulative genetic load using major susceptibility genes such as CFH (including the CFH R1210C variant), the ARMS2/HTRA1 locus, and the C3 K155Q variant. It also integrates minor variants across lipid metabolism and extracellular matrix pathways, including RAD51B, ABCA1, TIMP3, SLC16A8, and TGFBR1.
- Non-Genetic and Clinical Factors: These genetic risk markers are combined with traditional lifestyle and demographic metrics, specifically the patient's age, family history, smoking status, and Body Mass Index (BMI).
By aggregating these 21 combined predictors, clinical genomicists can highly accurately predict disease progression, achieving an Area Under the Curve (AUC) of 0.94 over 5 years and an AUC of 0.92 over 12 years
How does cigarette smoke accelerate epigenetic aging in the eye?
Cigarette smoke accelerates epigenetic aging in the eye by causing profound structural changes to the chromatin within retinal pigment epithelium (RPE) cells, directly shutting down their ability to repair themselves.
According to 2026 research from Johns Hopkins Medicine, exposing RPE cells to cigarette smoke condensate triggers this accelerated aging through three primary epigenetic mechanisms:
- Decreased Chromatin Accessibility: Smoke exposure significantly alters how DNA is physically packaged within the cell, reducing the physical accessibility of regions that contain essential genes for cellular survival and repair. This epigenetic change effectively "locks away" the cells' necessary repair pathways.
- Formation of Dysfunctional RPE Clusters: The acute stress from the smoke leads to the appearance of transcriptomically distinct, "dedifferentiated" clusters of RPE cells. In these affected cells, core functional genes that are essential for normal eye physiology (such as Slc6a20a and Myrip) are significantly downregulated.
- Loss of the Protective "Safety Net" in Aged Cells: The epigenetic damage from cigarette smoke is particularly lethal as eyes get older. While young RPE cells are able to survive acute smoke stress by transiently upregulating a protective subset of genes (involved in autophagy, mitochondrial quality control, and proteostasis), aged RPE cells fail to activate this response. This failure under stress leads directly to chromatin closure, a complete halt in cellular repair, and cell death.
Crucially, these smoke-induced epigenetic changes translate directly to human disease; researchers identified 1,698 specific gene expression changes shared between these smoke-exposed models and actual human donor RPE tissues from smokers and early-stage AMD patients. This demonstrates a highly conserved epigenetic mechanism where environmental stress like smoking accelerates eye aging, cell death, and ultimately exacerbates a patient's underlying genetic risk for macular degeneration
References
- Laine M, Jarva H, Seitsonen S, et al. Y402H polymorphism in complement factor H reduces binding to C-reactive protein. J Immunol. 2007;178(6):3831-3836.
- Toomey R, et al. The principal AMD-risk–associated CFH variant (Y402H) regulates lipoprotein levels in vivo. Proc Natl Acad Sci U S A. 2019;116(9):3718-3723.
- AMD Book. Complement Factor H (CFH). Available from: https://amdbook.org/content/complement-factor-h-cfh[Accessed June 2026].
- [13] Goldberg M, et al. The Complement Factor H (Y402H) risk polymorphism affects mitochondrial function and oxidative stress responses in iPSC-derived RPE. J Cell Sci. 2024;137(4):jcs261485.
- [9] Armento A, et al. Complement Factor H Y402H polymorphism and cigarette smoke extract modulate lipid pathways in stem-cell-derived retinal pigment epithelium. bioRxiv. 2026; doi:10.1101/2026.01.06.697989.
- [23] Geromedicine Review. Autophagy and mitophagy in retinal pigment epithelial homeostasis and age-related macular degeneration. Geromedicine. 2026;2026:0012.
- Golestaneh N, et al. Donor disease state and CFH genotype compromise mitochondrial function and elevate inflammation in iPSC-derived retinal pigment epithelium. J Trans Med. 2021;19(1):164.
- [24] RPE Mitophagy Review. Mitophagy pathways in retinal pigment epithelium under oxidative stress and in retinal diseases. Front Cell Dev Biol. 2023;11:1047714.
- Mitophagy in AMD. The role of mitophagy impairment in the pathogenesis of age-related macular degeneration. Int J Mol Sci. 2024;25(9):11105454.
- PINK1 Macular Study. Decreased PINK1-mediated mitophagy in perifoveal retinal pigment epithelium triggers epithelial-mesenchymal transition in early AMD. Autophagy. 2023;19(3):9980637.
- Genomic Analysis Consortium. Genetic analysis of age-related macular degeneration highlights precision therapy opportunities for patients with high polygenic risk. ResearchGate. 2024; doi:10.13140/RG.2.2.40284.4455.
- Sobrin L, et al. Composite risk score including family history and multi-locus genetic predictors separates high and low risk of AMD progression. Ophthalmology. 2024;131(11):11694608.
- [5] Polish AMD Study. Polygenic risk score stratification and genetic heritability in a Polish cohort with age-related macular degeneration. Hum Genomics. 2023;17(1):9821027.
- Updated PRS Performance. Performance of an updated polygenic risk score (PRS2023) for predicting late-stage age-related macular degeneration. Ophthalmology. 2024;131(2):189-197.
- GRADE Study Protocol. Genetic Risk Assessment of Degenerative Eye Disease (GRADE): prospective cohort study of polygenic risk scores. BMJ Open. 2023;13(10):e10594830.
- Protective Exome Study. Protective coding variants in CFH and PELI3 associated with a reduced risk of advanced age-related macular degeneration. Nat Genet. 2018;50(6):790-796.
- Systemic Complement Study. Risk alleles in CFH and ARMS2 are independently associated with systemic complement activation in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2011;52(13):9428-9434.
- [35] APOE Controversy. What do we know about APOE? Isoforms and clinical discrepancies. CureFFI. 2016; Available from: https://www.cureffi.org/2016/03/02/what-do-we-know-about-apoe/ [Accessed June 2026].
- [36] Baird PN, et al. The epsilon 2 and epsilon 4 alleles of the apolipoprotein E gene are associated with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2004;45(5):1311-1315.
- [1] APOE Structural Variants. Structural genetic variation in apolipoprotein E (APOE) and risk of age-related macular degeneration. JAMA Ophthalmol. 2022;140(12):1194-1202.
- Neuroscience News. Cigarette smoke triggers epigenetic age acceleration in the eyes. Neuroscience News. 2026; Available from: https://neurosciencenews.com/smoking-eye-aging-epigenetics-30208/ [Accessed June 2026].
- Ocular Epigenetics. Epigenetic modifications and gene-environment interactions in age-related macular degeneration. Front Med. 2024;11:12347735.
- Johns Hopkins Medicine. Cigarette smoke accelerates eye aging via epigenetic changes, Johns Hopkins study finds. JHM Press Release. 2026; Available from: https://www.hopkinsmedicine.org/news/newsroom/news-releases/2026/02/cigarette-smoke-accelerates-eye-aging-via-epigenetic-changes-johns-hopkins-medicine-study-finds [Accessed June 2026].
- [8] EYE-RISK Consortium. Long-term dietary zinc and regulatory pathways in slowing AMD progression: results from the EYE-RISK project. EYE-RISK Portal. 2020; Available from: https://www.eyerisk.eu/news-events.html[Accessed June 2026].
- Thee EF, Acar IE, Colijn JM, et al. Systemic metabolomics in a framework of genetics and lifestyle in age-related macular degeneration. Metabolites. 2023;13(6):701.
- [16] Wet AMD Progression. The ARMS2-HTRA1 locus, cigarette smoking, and oxidation-induced inflammatory responses in wet AMD. Front Med. 2021;8:8133762.
- Mitophagy Enhancers. Autophagy and mitophagy modulation as potential disease-modifying therapies in age-related macular degeneration. Pharmacol Res. 2025;203:12938228.
- HTRA1 Overexpression. Overexpression of HTRA1 leads to ultrastructural changes in the elastic layer of Bruch's membrane in transgenic mice. PLoS One. 2011;6(7):e31490.
- ARMS2-HTRA1 Locus Controversy. Deciphering the causal gene at the 10q26 locus in age-related macular degeneration. Prog Retin Eye Res. 2024;99:12053538.
- [21] HTRA1 Substrates. Overexpression of HTRA1 in mice compromises Bruch's membrane integrity and degrades fibulin-5 and ApoE. Invest Ophthalmol Vis Sci. 2019;60(9):1233.
- [42] NCBI Gene. CFH complement factor H [Human (Homo sapiens)]. NCBI Gene Database. Available from: https://www.ncbi.nlm.nih.gov/gene/3075 [Accessed June 2026].
- CFH Risk and Protection. Complement Factor H gene variants and protective haplotypes in Caucasian populations. AMD Book. 2021; Available from: https://amdbook.org/content/complement-factor-h-cfh [Accessed June 2026].
- Rare Coding Variants. PELI3 and CFH protective low-frequency variants in advanced age-related macular degeneration. Hum Mol Genet. 2018;27(14):2511-2518.
- [33] Exome BeadChip Study. Meta-analyses of exome array data identify protective variants in PELI3 and CFH for advanced AMD. Am J Hum Genet. 2017;100(1):155-162.
- Complement Concentrations. Alternative complement activation and concentrations of C3d/C3 ratio in carriers of CFB and CFH protective alleles. Ophthalmology. 2011;118(12):2411-2418.
- Lipid and ECM Pathways. Detailed molecular mechanisms of ApoE, ABCA1, TIMP3, and other minor lipid/ECM variants in AMD pathobiology. Front Aging Neurosci. 2024;16:11274963.
- [43] ABCA1 Copenhagen Study. Genetic variants in ABCA1, HDL cholesterol concentrations, and risk of age-related macular degeneration in the general population. Eur J Epidemiol. 2023;38(10):10501952.
- Cholesterol-Related Genes. Susceptibility of ABCA1, CETP, and LIPC polymorphisms with lipid levels and degenerative diseases. Neurobiol Aging. 2013;34(3):1024-1031.
- [2] ABCA1 Pathogenesis. ATP-binding cassette transporter A1 (ABCA1) and risk of neovascular and non-neovascular AMD. Copenhagen Gen Pop Study. 2023; doi:10.1007/s10654-023-01021-4.
- [29] RPE Lipid Pathobiology. Dysregulated lipid transport and metabolism in the retinal pigment epithelium in AMD. J Lipid Res. 2022;63(2):8817708.
- [44] Thee Systemic Metabolomics. Systemic metabolomics in a framework of genetics and lifestyle: the EYE-RISK Project. Metabolites. 2023;13(6):701. doi:10.3390/metabo13060701.
- [45] EYE-RISK Publications. Dietary intake of vegetables, fruit, and fish is beneficial for slowing AMD progression. Am J Ophthalmol. 2019;198:70-79.
- Metabolomics Review. Stage-specific plasma metabolites associated with early-intermediate and late age-related macular degeneration. MedPage Today. 2023; Available from: https://www.medpagetoday.com/resource-centers/wet-amd-current-perspectives/age-related-macular-degeneration-circulating-metabolites-genetics-and-lifestyle/4747 [Accessed June 2026].
- [46] Rotterdam Study. Systemic Metabolomics in a Framework of Genetics and Lifestyle in Age-Related Macular Degeneration. Erasmus University Rotterdam. 2023; Available from: https://pure.eur.nl/en/publications/systemic-metabolomics-in-a-framework-of-genetics-and-lifestyle-in/ [Accessed June 2026].
- [47] Moor AE, Moor G. Fragment-sequencing and single-cell resolution metabolomics in tissues. Nature Communications. 2023;14:7775.
- Handa JT, et al. Cigarette smoke accelerates eye aging via epigenetic changes. PNAS. 2026;123(3):e2505412123.
- RPE Heterogeneity. Intravitreal cigarette smoke condensate and aging induce transcriptomically distinct RPE clusters in mice. PNAS. 2026;123(3):e2505412123.
- [48] Futurity Eye Study. How cigarette smoke rewires RPE chromatin structure and accelerates eye aging. Futurity. 2026; Available from: https://www.futurity.org/cigarette-smoke-eye-aging-3327012/ [Accessed June 2026].
- [40] Earth Science News. Smoking rewires eye cells, speeding up vision loss and degeneration. Earth.com. 2026; Available from: https://www.earth.com/news/smoking-rewires-eye-cells-speeding-up-vision-loss-and-degeneration/ [Accessed June 2026].
- Epigenetic Sabotage. Cigarette smoke triggers epigenetic age acceleration and chromatin closure in RPE cells. Neuroscience News. 2026; Available from: https://neurosciencenews.com/smoking-eye-aging-epigenetics-30208/[Accessed June 2026].
- ARMS2 Causality Doubt. Analysis of the R38X nonsense mutation rs2736911 and heritability patterns at the 10q26 locus. PLoS Genet. 2018;14(4):e1007353.
- [18] North Indian Cohort. Genotype-phenotype correlations for ARMS2, HTRA1, and CFH SNPs in neovascular age-related macular degeneration. Ophthalmic Genet. 2024;45(2):12947101.
- Multi-Ethnic Locus Study. Minor allele frequencies and disease risk of ARMS2/HTRA1 locus in Asian versus Western populations. J Hum Genet. 2024;69(5):11717327.
- CRISPR ARMS2 Study. CRISPR editing of rs10490924 demonstrates mitochondrial ROS elevation and attenuated SOD activity in iPSC-derived RPE. Proc Natl Acad Sci U S A. 2023;120(22):e10175836.
- HTRA1 Knockout Phenotype. Ablation of Htra1 leads to progressive cone and rod photoreceptor dysfunction and Bruch's membrane deposits. Invest Ophthalmol Vis Sci. 2025;66(3):11948579.
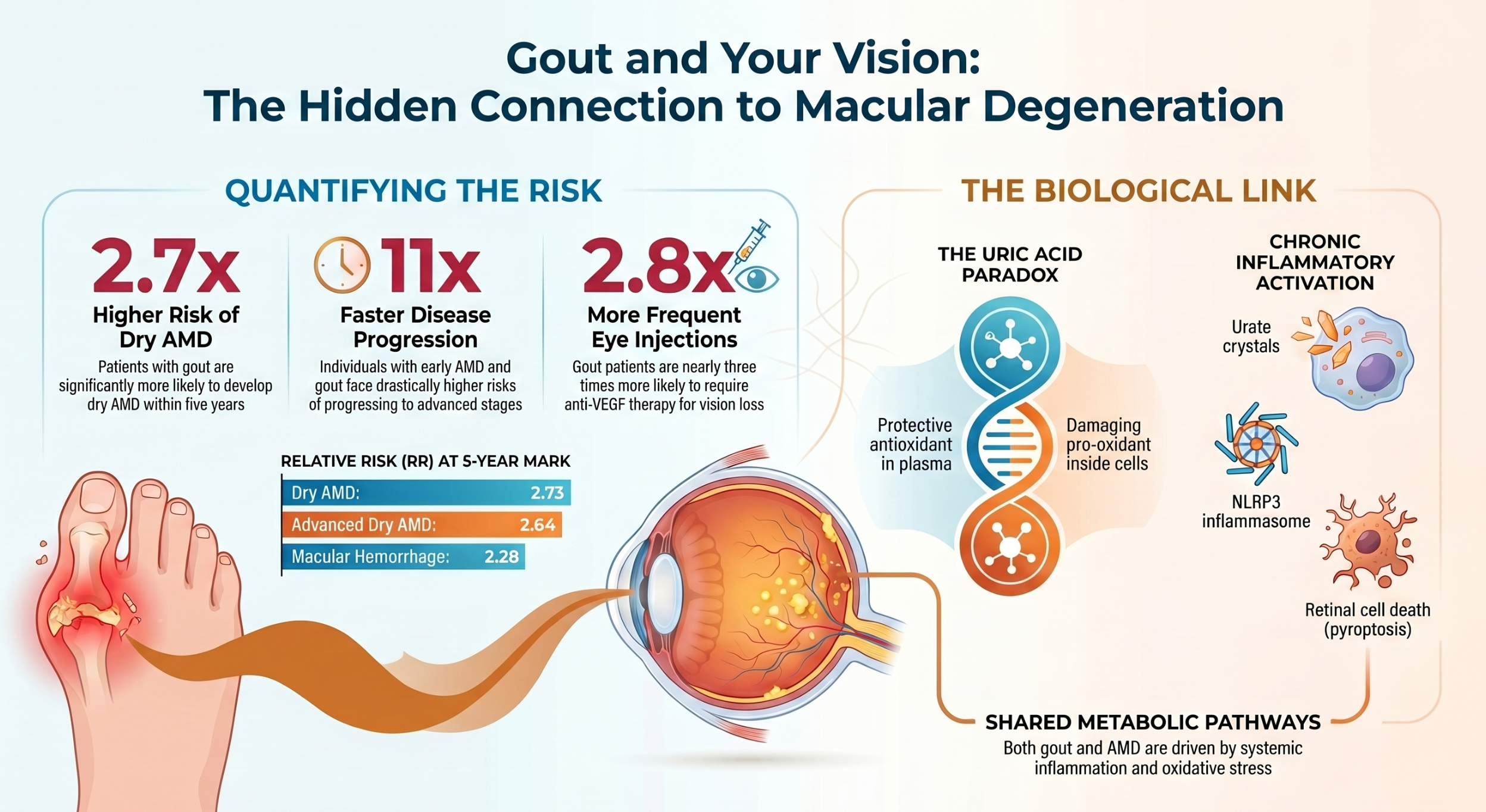
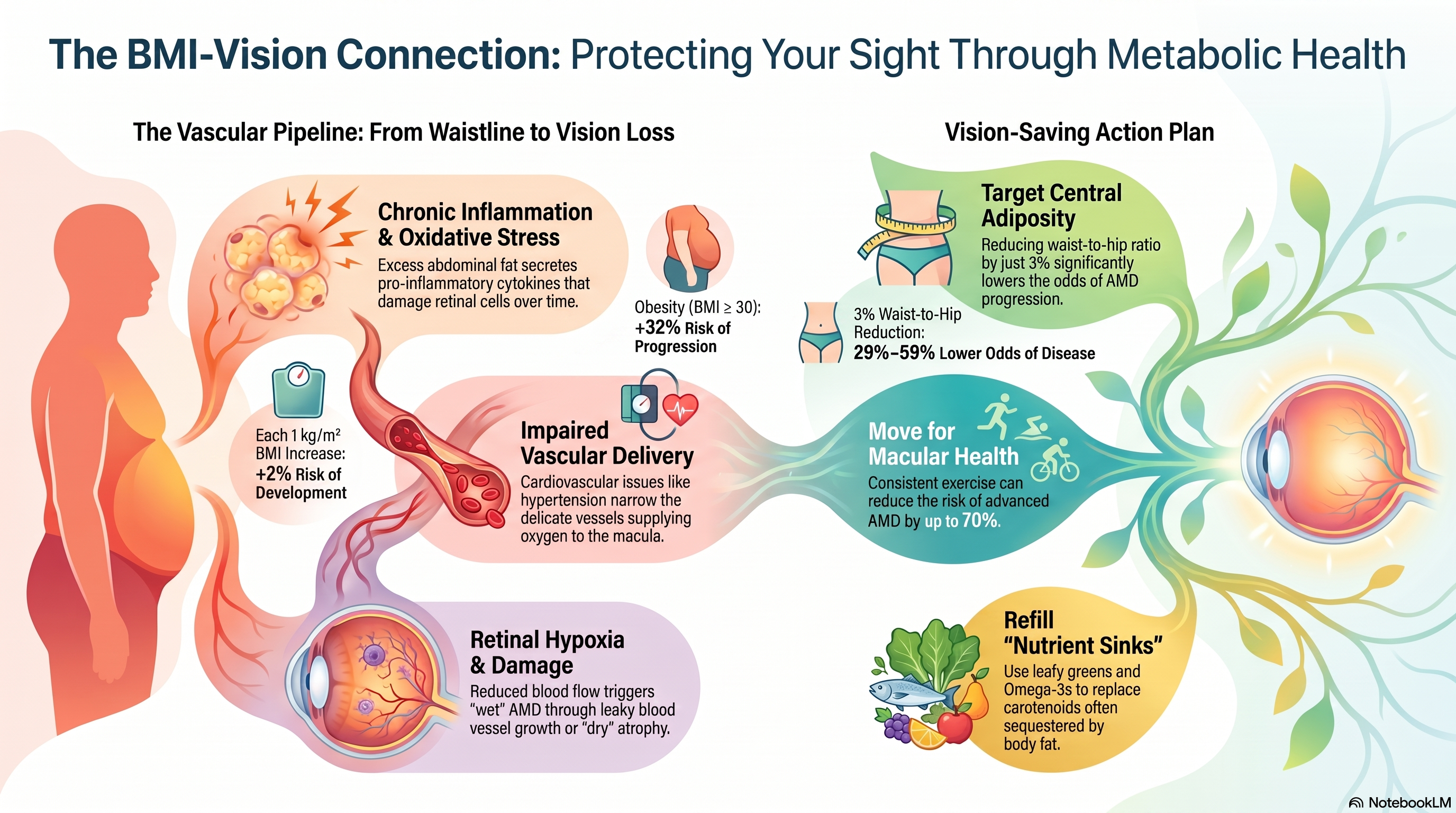
{kind=link}
Leave a comment
This site is protected by hCaptcha and the hCaptcha Privacy Policy and Terms of Service apply.